| Title | HUMAN THIOREDOXIN ADULT T CELL LEUKEMIA DERIVED FACTOR ACTIVATES THE ENHANCER BINDING PROTEIN |
|---|---|
| Year | |
| Author | Takashi Okamoto, Hiroyuki Ogiwara, Takuma Hayashi, Akira Mitsui, Takumi Kawabe, Junji Yodoi |
| Publisher |
Human thioredoxin/adult T cell leukemia derived factor activates the enhancer binding protein of human immunodeficiency virus type 1 by thiol redox control mechanism
Takashi Okamoto, Hiroyuki Ogiwara3, Takuma Hayashi, Akira Mitsui1, Takumi Kawabe2, and Junji Yodoi2
Virology Division, National Cancer Center Research Institute, Tsukiji 5-1-1, Chuo-ku, Tokyo 104, Japan
1Basic Research Laboratory, Ajinomoto Co., 1-1 Suzuki-cho, Kawasaki 210, Japan 2Virus Research Institute, Kyoto University, Shogoin-Kawaharacho, Sakyo, Kyoto 606, Japan 3Present address: 2nd Department of Surgery, Hamamatsu Medical College, Shizuoka 431-31, Japan
Key words: nuclear factor kappa B, DNA binding protein, signal transduction, acquired immunodeficiency syndrome, adult T cell leukemia, interleukin 2 receptor α
Abstract
Transcription from the human immunodeficiency virus type 1 (HIV-1) provirus is activated by a cellular factor, NFxB, recognizing the tandemly repeated 10-base-pair sequences, termed the xB sequence, present in the enhancer region within the viral long terminal repeat (LTR). Using electrophoretic mobility shift assay (EMSA), which demonstrates specific DNA- protein interaction In vitro, we could demonstrate that reducto-oxidative modulation of NFxB dramatically changes its DNA binding activity and that a cellular physiological reducing catalyst, thioredoxin (TRX) also known as adult T cell leukemia derived factor (ADF), fully restored the DNA-binding activity of the oxidized NFxB. We also observed that purified TRX/ADF protein could augment gene expression from HIV L TR as demonstrated by transient chloramphenicol acetyltransferase (CAT) assay. These observations confirmed the previous notion that ADF might be an inducing factor of cellular interleukin-2 receptor α subunit (IL-2R α) through the xB sequence that is a common central cis-regulatory element in both IL-2Rα and HIV gene expression. These observations indicate that reducto-oxidative regulation (or redox regulation) of a cysteine residue(s) on the NFxB molecule might play an important role in its specific DNA interaction and that it might provide a clue to the understanding of a pathway of cellular signal transduction to NFxB that is independent from the known pathways involving protein phosphorylation.
Introduction
NFxB is a pleiotropic mediator of transcription for various promoters (1 – 8). It activates a wide variety of viral and cellular genes including human immuno-deficiency virus-1 (HIV-1) (9 -13), interleukin-2 receptor α (IL-2R α) (4, 14, 15), tumor necrosis factor- α (TNF- α) (16) and GM-CSF (17). NFxB is initially located in the cytoplasm as an inactive form associated with an inhibitory protein, lxB (18). The finding that phorbor esters induce nuclear translocation of NFxB, and that NFxB is released in vitro by treatment of the cytosol fraction with purified protein kinase C (PKC) (19), have suggested that lxB may be inactivated by phosphorylation. However, one of the few known physiological inducers of NFxB, TNF- α, was found to activate NFxB even in the presence of protein kinase inhibitors, implying that a kinase independent mechanism was involved (20).
Within the HIV-1 long terminal repeat (LTR), two tandemly repeated xB sites have been identified in the region initially identified as enhancer. This cis-regulatory element has been shown to regulate the transcriptional inducibility of HIV in activated T cells and in TNF-treated cells (2,9,12,20,21 ). Thus, NFxB may serve as an intracellular mediator in activation of the latent HIV-1 provirus and thus participate in a critical step preceding the clinical manifestation of the acquired immunodeficiency syndrome (AIDS).
In T cells transformed by human T lymphotropic virus type 1 (HTLV-1), NFxB is constitutively activated causing overexpression of IL-2R α on the cell surface (15,22) and release of TNF- α (16) and GM-CSF (17). Moreover, replication of HIV-1 is greatly augmented in these cells (23,24) and its augmentation is dependent on the xB sites within LTR (9,12,21). Interestingly, Tagaya et al. (25) demonstrated that a cellular factor, known as adult T cell leukemia-derived factor (ADF), which is released from cells transformed by HTLV-1 (26,27) or Epstein- Barr virus (28) and has activity to stimulate IL-2R α expression, is the human homolog of thioredoxin (TAX) (see ref. 29 for review). Human TAX is a 13 kDa thiol protein that is a strong catalyst of dithioldisulfide exchange reactions (A. Mitsui, T. Hirakawa, and J. Yodoi, submitted). TRX participates in redox reactions through reversible oxidation of its active center dithiol to a disulfide. The actual role of TRX in inducing IL-2Rα is to be clarified, but there is much evidence to suggest that the reducto-oxidative conditions of sulfhydryl (SH) groups of some nucleic acid-binding proteins reversibly modulate their binding capabilities (30-33). Recently, several reports have indicated that reducto-oxidative conditions modulated the DNA binding activity of NFxB by using non-physiological inorganic reagents (34-36). These findings prompted us to investigate whether redox regulation by the TRX system participates in the regulation of HIV gene expression.
Methods
Nucleic acids
Oligodeoxynucleotides were synthesized in an Applied Biosystems 381 DNA synthesizer (Applied Biosystems. USA).
Preparation of nuclear and cytosolic extracts
Nuclear and cytosolic (S100) extracts were prepared from MT2 cells (37) in the logarithmic growth phase by the method of Dignam et al. (38). NFxB-activity in the crude MT2 nuclear extract was partially purified through heparin- agarose and DEAE-Sepharose columns as reported previously (39,40). All operations after cell harvest were performed at 4°C. All buffers contained protease inhibitors. 0.5 mM phenylmethylsulfonyl flouride (PMSF) and 0.01 U/ml aprotinin. Briefly, the crude nuclear extract after dialysis against a buffer D (containing 20 mM Tris- HCI (pH 7.9), 0.1 M KCI, 0.2 mM EDTA, 1 mM DTT and 20% glycerol) was loaded onto a heparin- agarose column, washed with five column volumes of buffer D, and the bound proteins were eluted with buffer D with 0.4 M KCI. This 0.4 M KCI eluate was dialysed against buffer D and loaded onto a DEAE- Sepharose column. The NFxB activity recovered in the 0.225 M KCI eluate (called ‘DE0.225’) was concentrated by ultrafiltration (using Centriprep; Amicon, MA, USA) and dialysed against buffer containing 20 mM HEPES-KOH (pH 7.9), 0.1 M KCI, 0.2 mM EDTA, 0.5 mM PMSF, and 20% (v/v) glycerol (no DTT was added) and stored in liquid nitrogen until use. The endogenous TAX, examined by Western blotting with specific antisera, and its activity, measured by the ability to convert NADPH to NADP+, was not detected in the MT2 DE0.225 fraction.
DNA binding assay
Kinase reactions of oligonucleotide DNA were performed with [-γ-32P] ATP and T4 polynucleotide kinase. Binding reactions were performed at 30°C for 10 min in a total volume of 10 µl of buffer containing 20 mM HEPES-KOH (pH 7.9), 60 mM KCI , 1 mM MgCI2, 5% (v/v) glycerol, 0.1% NP40, 1.0 µg of poly(dldC), 0.1 ng (20 000-30 000 c.p.m.) of labeled probe, and DE0.225 containing NFxB, DTT was not added in the experiments using sulfhydryl-modifying reagents. After addition of loading buffer, the mixture was analyzed by electrophoresis in 6% nondenaturing polyacrylamide gel with 0.5 x TBE buffer (4.5 mM Tris, 4.5 mM boric acid, 0.1 mM EDTA, pH 8.0). For competition assays, the competitor DNA (5 ng; i.e. 50-fold excess over the probe) together with poly(dl-dC) was preincubated with NFxB on ice for 5 min before addition of the rest of the reaction mixture. NFxB usually gave two major retarded bands on electrophoretic mobility shift assay (EMSA), as reported previously (3,7;8,14,21 ,41 ). The additions of the wild-type xB sequences reduced the intensities of both bands equally.
Modification of sulfhydryls
N-ethylmaleimide (NEM) and diamide [diazenedicarboxylic acid bis(N,N-dimethylamide)] were purchased from Sigma (St Louis, USA). NEM alkylates and irreversibly blocks free SH groups by the following reaction:
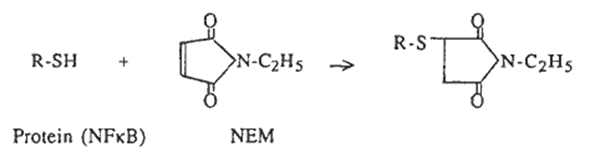
On the other hand, diamide reversibly converts thiols to disulfides (42) and the diamide-oxidized protein could be recovered by appropriate reducing agents such as DTT, 2-mercaptoethanol (2-ME) and the reduced form of TRX.
Recombinant human TRX. or ADF, and its mutant were purified to near homogeneity, reduced by incubation with excess DTT (usually 10 mM OTT was used for reducing 1 mM TRX) for 1 h at room temperature and dialyzed overnight against buffer saturated with nitrogen gas. Purified and reduced TRX was stored in liquid nitrogen until use.
Transfection and chloramphenicol acetyltransferase (CAT) enzyme assay
For transfection of plasmid DNA into COS-1 cells, the calcium phosphate precipitation protocol was used as previously reported (9,24). Construction of HIV LTR- CAT, CD12, and its deletion mutant, CD52, lacking the upstream sequence from nucleotide position – 65, was reported in the previous papers (13,40). Cells were harvested after 48 h of transfection and the CAT enzyme activity in 100 µg of cell lysate was measured by the standard method (9,24). Purified recombinant TRX/ADF was added to the cell culture media 24 h before cell harvest at the concentrations indicated. Protein concentration of each lysate was determined using Bradford’s method (BioRad. USA).
Results
Preparation of ‘activated’ NFxB and its specificity
NFxB was partially purified from one of the HTLV-1-transformed cell lines, MT2 (37). For this, a crude nuclear extract was prepared from MT2 cells in the logarithmic growth phase by the method of Dignam et al., (38) and NFxB in the extract was fractionated using heparin- agarose and DEAE- Sepharose columns (39,40) (Fig. 1). Since in MT2 cells genes regulated through the xB motif, such as IL-2Rα and GM-CSF, are constitutively activated like other HTLV-1-immortalized cell lines, the NFxB protein prepared from the MT2 nuclei are considered to be fully competent for specific DNA recognition. Oligonucleotide DNAs containing xB sequences of the HIV-1 (2), human IL-2Rα (3), and mouse class 1 major histocompatibility complex (MHC) (41) genes and their mutants were synthesized chemically for use as probes or competitors (Fig. 2). By EMSA (2), xB-binding activity was demonstrated in the fraction of nuclear extract of MT2 cells eluted from heparin- agarose with 0.4 M KCI and from DEAE- Sepharose with 0.225 M KCI (Fig. 1b, lanes 4 and 7). This preparation yielded – 40 fold purification of the xB-binding protein from the crude nuclear extract. A cytosol fraction, S100, was prepared from MOLT 4 cells, a human CD4 cell line. When the S100 was detergent treated with deoxycholate and NP40, xB-binding activity was detected by EMSA (lanes 9 and 10) as previously shown for the NFxB-IxB complex (18). It was also noted that the mobility of the shifted band detected with the S100 fraction was similar to the band detected with the nuclear fraction in the presence of the detergents (lanes 8 and 9). A slight difference in mobility of the NFxB-DNA complex was noted in lane 8, as compared with that in lane 7, which was probably due to the presence of detergents.
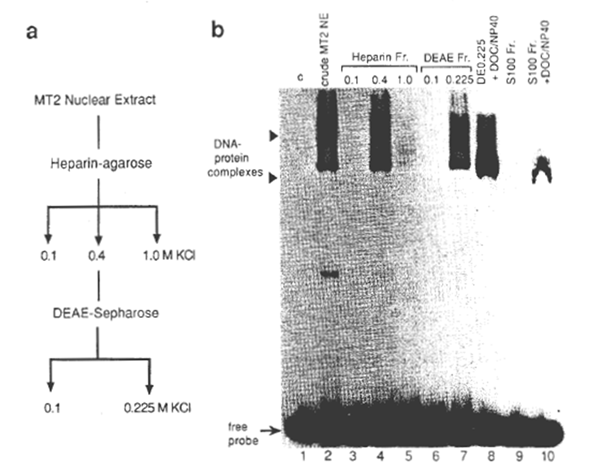
Fig. 1. (a) Scheme for the preparation of the HIV-1-enhancer (xB sequence)-binding protein. The crude nuclear extract (NE) was prepared from MT2 cells and fractionated as described in Methods. (b) The xB DNA-binding activities of fractions after each stage of purification by EMSA. 32PIabeled double-stranded HIV wt probe (see Fig. 2a for the sequence) was incubated with each fraction. The binding reactions and electrophoresis were as described under Methods. One microliter (about 106 cell equivalent) of each fraction was added to the binding reaction (lanes 2 through 7). Two retarded bands are indicated by close arrow heads. In lane 1, no protein was added (c, control). In lanes 8 and 10, 2 µl of DEAE 0.225 nuclear fraction or cytosolic fraction of MT2 cells (S100) were incubated with 0.8% deoxycholate and 1.2% NP40 (final concentrations) before addition of the probe. The attenuation of the upper band and the slight downward shift of the lower band with the DE0.225 fraction was noted upon treatment with detergents, probably due to the dissociation of the protein complexes or the conformational changes. Positions of DNA- protein complexes and free unbound probe are shown by arrow heads and an arrow, respectively
To examine further the specificity of NFxB activity, various oligonucleotides (Fig. 2a) were synthesized for cold competition experiments. As shown in Fig. 2b, NFxB-binding to the HIV-1 xB probe was prevented by addition of 50-fold molar excess of the unlabeled NFxB oligonucleotide ‘HIV wt’, ‘IL-2R wt’, or ‘MHC-1 wt’ to the reaction mixture, but not by addition of the mutant oligonucleotide ‘HIV mut’, ‘IL2R mut’, or ‘MHC-1 mut’ (Fig. 2b). Consistent with these findings, the labeled probes of the mutant DNAs were not recognized by NFxB (not shown). From these observations as well as the similarities of the gel retardation pattern with previous reports (also showing double retarded bands), we concluded that the DNA-binding activity is due to the activity of NFxB.
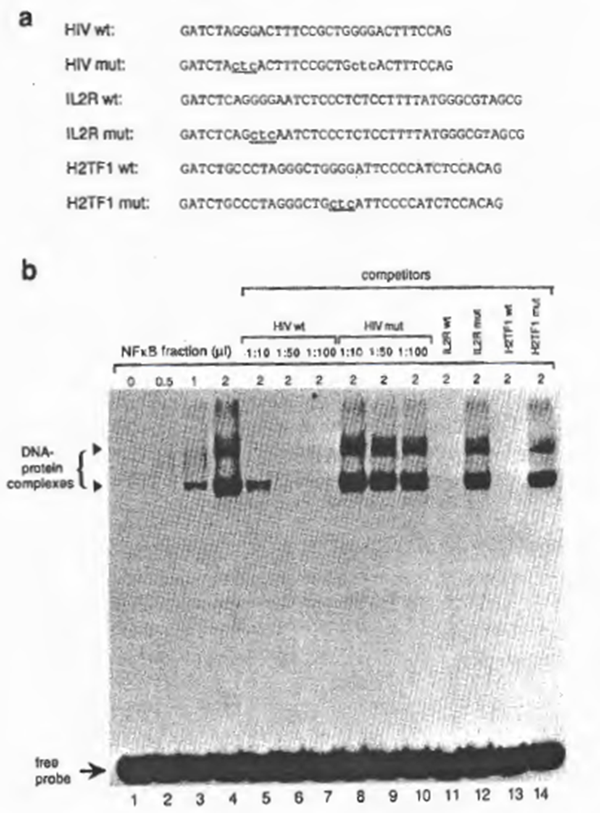
Fig. 2. (a) DNA sequences of synthetic oligonucleotides for xB binding sites used for EMSA. Indicated are the HIV-1 enhancer elements located between -104 and -78, containing the wild-type xB sites (‘HIV wt’), the IL2-Rα enhancer region containing a single xB site (nucleotide position from -269 to – 235) (‘IL2R wt’), and their mutants, ‘HIV mut’ and ‘IL2R mut’, respectively. Mutations are indicated by lower-case letters with underlines. The linker sequences (GATCT in the 5′ end and G in the 3′ end) were added for cloning purposes. (b) Analysis of binding to the wild-type and mutant NFxB binding sites by EMSA. 32P-Iabeled HIV wt double-stranded oligonucleotide was incubated with partially purified NF xB prepared from MT2 (DE0.225 nuclear fraction). Dose-dependent binding was observed in lanes 2 through 4. The specificity of the protein- DNA interaction is demonstrated in lanes 5 through 11. In lanes 6 through 8, increasing amounts of cold HIV wt DNA were added (molar ratios of the probe to the competitor are indicated). In lanes 9 to 11 , the unlabeled competitors, IL-2R wt, HIV mut, or IL-2R mut, were added at a molar ratio of 1:50.
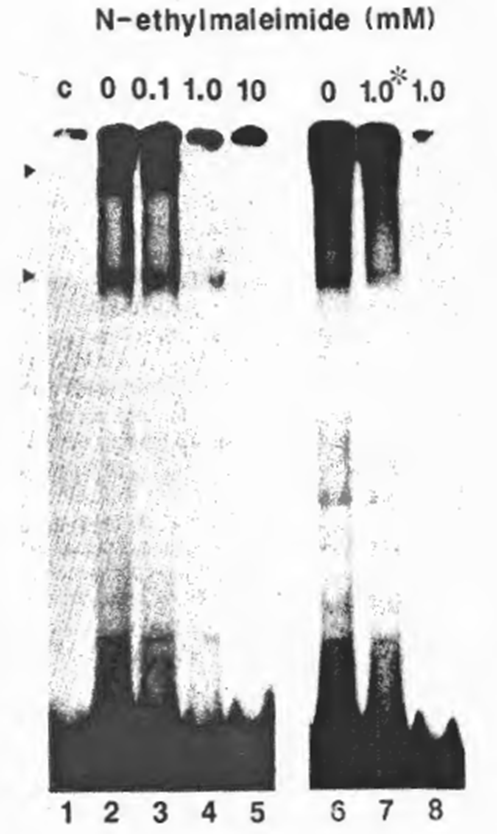
Fig. 3. Requirement of free SH groups for DNA-binding activity of NF xB. The partially purified NFxB was treated with NEM at a final concentration of 0.1 (lane 3), 1.0 (lane 4), or 10 mM (lane 5) on ice for 5 min before the EMSA reaction with the HIV wt probe. Lanes 6-8 show the effect of DNA-binding on susceptibility to NEM: for Lane 7, NEM (1.0 mM final concentration) was added after incubation with the probe; for lane 8, NFxB was treated with NEM before the binding reaction as in lane 4, and for lane 6, no NEM was added. No DTT was added in the reaction to avoid possible interaction with NEM.
Involvement of sulfhydryls on the NF xB molecule in DNA-binding
The requirement of free sulfhydryl groups of NFxB in DNA recognition was investigated using a moderate sulfhydryl-specific alkylating agent, NEM, to block the sulfhydryls on the molecular surface irreversibly. Pretreatment of the MT2 nuclear extract fraction (DE0.225) containing ‘activated’ NFxB with 1 mM NEM before addition of the HIV-1 xB probe prevented formation of a DNA- protein complex, as shown by EMSA (Fig. 3), whereas addition of the same concentration of NEM after DNA- protein interaction did not prevent the binding (Fig. 2, lane 7). These results indicate that NEM-mediated alkylation involves only the outer sulfhydryls on the NFxB molecule and that the blocking of DNA – protein interaction by NEM was not due to a drastic conformational change.
Redox regulation of NFxB activity in vitro
Next we examined the effect of diamide, which catalyzes chemical oxidation of free sulfhydryls, and of purified recombinant human TRX, a physiological reducing catalyst, on DNA recognition by NFxB. In the following experiments, DTT was not added to most of the reactions because of its possible intervention with the oxide-reductive reagents to be examined, although the clarity of the retarded bands deteriorated (which was probably due to partial oxidation of the protein). As demonstrated in Fig. 4b, oxidation of NFxB by diamide inhibited the binding dose dependently (lanes 1 through 6). The aberrant band obtained on treatment with 1 mM diamide (lane 5) did not appear to be specific for the xB sequence judging from the results of competition experiments (compare lanes 5, 7, and 8), and so was probably due either to modification of the NFxB protein by oxidation, such as by a conformational alteration or dissociation of the multimer structure, or to the formation on oxidation of a non-specific DNA-binding protein that was co-fractionated with NFxB. Reduction of oxidized NFxB by recombinant human TRX or 2-ME restored the binding activity of NFxB. Considering the concentrations of these reducing reagents which were required to restore the NFxB, TRX appeared to have nearly 1000-fold higher efficiency than 2-ME. However, a mutant human TRX with only a single amino acid replacement of cysteine 31 by serine (Fig. 4a), resulting in loss of the protein-reducing activity as shown by insulin disulfide-reducing assay ((43); Misui et al., submitted), did not counteract the effect of diamide (Fig. 4b, lanes 9 through 15).
Similar oxidation- reduction experiments were done with other transcription factors, and representative results obtained with CCAAT-binding protein (CBP) are shown in Fig. 5. Oxidation and reduction of the partially purified CBP were performed as for Fig. 4b. The DNA binding activity of CBP was not affected by treatment with diamide even at a concentration of 4 mM (lane 5) or by further treatment with TRX (lanes 6 through 8). Treatment with reducing agents only also did not change the binding activity
(not shown).
Reduction of NFxB by human TRX appeared to be temperature dependent: the binding activity was restored by treatment of oxidized NFxB with diamide for 5 min at 30°C, but not at 4°C (Fig. 6a). However, the binding activity was partially restored by prolonged incubation of oxidized NFxB with human TRX at 4°C (not shown). This temperature dependence of reduction was in contrast with that by the inorganic reducing agent 2-ME, which restored the binding activity equally at both temperatures although the reducing activity of 2-ME in this assay system was -1000 x less than that of human TRX. Mutant TRX could not restore the NFxB activity at either temperature, confirming that cysteine 31 was crucial for the reducing activity (lanes 4 and 8). These observations demonstrated that TRX reduced the oxidized NFxB and restored its DNA-
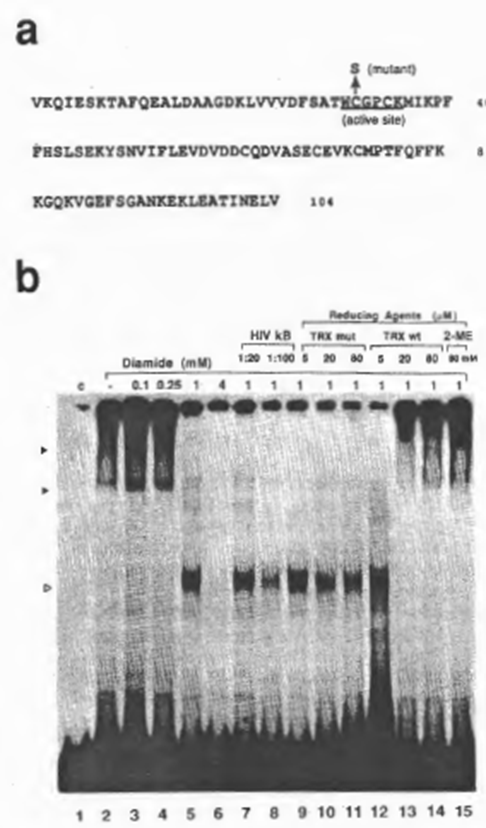
Fig. 4. Reducto-oxidative regulation of the DNA-binding activity of NFxB. (a) Amino acid sequences of human TRX/ADF (25). The active site, which is completely conserved in human and E. coli, is underlined.
A mutant in which cysteine 31 was replaced by serine had no ability to catalyze the dithiol-disulfide exchange reaction, as shown by insulin reduction assay (43) (Mitsui et al., in preparation). (b) Reversible inactivation of NFxB activity by oxidation and reactivation catalyzed by TRX. EMSA reactions were performed as for Figs 1 and 2 except that 1 mM (final concentration) DTT was not added to the reaction mixture. Incubation of NFxB with diamide was performed on ice for 5 min and the subsequent reduction with TRX at the final concentrations indicated was carried out at 30°C for 5 min. For lanes 3-6, increasing concentrations of diamide were added. The abberrant band (indicated by an open arrow head) observed in lane 5 (with 1 mM diamide) appeared not to be specific for the xB sequence judging from the results of competition experiments with cold DNA (lanes 7 and 8). Diamide pretreated NFxB was subsequently incubated with wild-type human TRX (TRX wt) (lanes 9- 11) or the mutant (TRX mut) (lanes 12- 14). In lane 15, NFxB oxidized with diamide was treated with 2-mercaptoethanol (2-ME) (80 mM final concentration).

Fig. 5. Effects of oxidation and reduction of CCAAT binding protein (CBP) on its DNA-binding activity. The 0.1 M KCI eluate from the DEAE- Sepharose column (Fig. 1) was used as a source of CBP. The probe for CBP was the synthetic oligonucleotide, 5′-GATCCAAACCAGCCAATGAGAACTGCTCCA-3′ (44).
binding activity through its active thioi on cysteine 31 by the enzymatic mechanism. Preincubation of TRX with diamide before incubation with NFxB was less efficient than addition of diamide after preincubation of TRX with NFxB in restoring the NFxB activity (Fig. 6b), suggesting that TRX-responsive sulfhydryls on the NFxB molecule require a highly reduced form of TAX (in which the active sulfhydryls are probably most prone to be oxidized) or that TRX in the presence of its target protein is more resistant to oxidation as in the case of the TRX-phage T7 DNA polymerase complex (45).
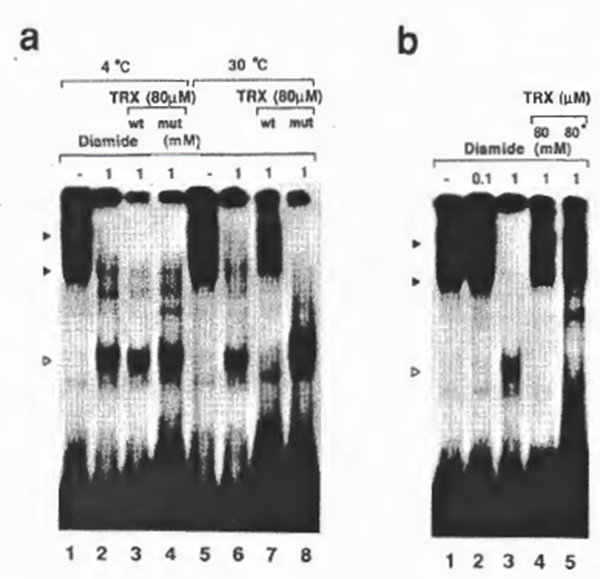
Fig. 6. (a) Temperature dependence of the action of TRX. For lanes 6-8, protein reduction by TRX was performed at 30°C tor 5 min, while for lanes 2 and 3, the reaction was carried out at 4°C for 5 min. Lanes 3 and 7, mutant TRX; lanes 4 and 8, wild-type TRX. Temperature dependence of TRX activity has also been observed by insulin disulfide reducing assay (according to the method of Slaby and Holmgren (45); also our unpublished results at 12°C and 30°C). (b) Effect of preincubation of TRX with NFxB on its susceptibility to oxidation by diamide. For lanes 1-4, the reaction protocol was as for Fig. 3b. For lane 5, the same amount of TRX as tor lane 4 was preincubated with diamide on ice for 5 min before its incubation with NFxB. Autoradiographs of -5-day exposure are shown to demonstrate the differences of DNA binding.
Effect of TRX/ADF on HIV gene expression in vivo
To examine further the effect of TRX/ ADF on HIV gene expression in vivo, a transient CAT assay was performed with COS-1, a monkey kidney-derived cell line, and CAT gene expressing plasmids under the control of HIV-1 LTR, CD12, or its derivative, CD52, with the deleted upstream sequence from – 65, therefore not containing the xB sequences (13,40). Plasmids expressing CAT under the control of chicken β-actin promoter were used as control. After transfection, purified recombinant TRX/ADF was added 24 h prior to cell harvest and CAT enzyme activities were measured according to the standard protocol (9,24). Representative results are shown in Fig. 7. When cells were treated with TRX/ADF, CAT enzyme activity from the wild-type HIV LTR was stimulated in a dose-dependent manner. However, the HIV LTR mutant lacking the upstream sequence including xB sites, as well as the control promoter, did not respond to the effect of TRX/ADF. These observations with the cell culture experiment confirmed the in vitro effect of TRX/ADF on the DNA-binding activity of NFxB.
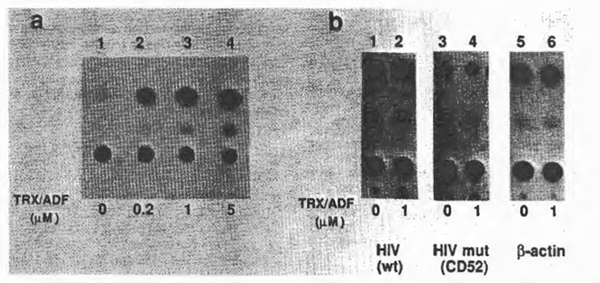
Fig. 7. Effects of purified TRX/ADF on HIV gene expression in vivo. Results of transient CAT gene expression assays were demonstrated. (a) Dose dependent stimulation of HIV gene expression by purified recombinant TRX/ADF. A CAT-expressing plasmid under the control of HIV LTR, CD12. was transfected into COS cells. Purified TRX/ADF was added to the cell culture medium to the indicated concentrations 24 h before cell harvest. (b) Effect of delet1on of the upstream element containing the NFxB binding sites from the HIV LTR. A deletion mutant lacking the upstream sequence (up to the nucleotide position -65 from the CAP Site), CD52, was Similarly tested for 1ts responsiveness to the exogenous TRX/ADF. A CAT-expressing plasmid under the control of chicken β-actin promoter (46) was also used as a control.
Discussion
Here we present evidence which suggests that TRX/ADF might activate HIV gene expression by a dithiol-disulfide exchange reaction of the cysteine residues on the NFxB molecule. First, the present study has shown that free SH groups of NFxB required for its specific DNA-binding activity. Oxidation of already ‘activated’ NFxB prepared from MT2 cell nuclei inactivated its specific DNA-binding activity and subsequent reduction of the oxidized protein by TRX/ADF efficiently restored its activity. Moreover, the transient CAT gene expression assay has demonstrated that gene expression from HIV LTR was augmented by the exogenously added TRX/ADF. Since this activity was abolished when the upstream cis,regulatory region containing the NFxB-binding sites was deleted from the HIV LTR, this in vivo effect of TRX/ADF might also be mediated through NFxB.
This kind of control mechanism involving sulfhydryls of cysteine residues is collectively called ‘redox regulation’ (29). Similar phenomena have been observed with other nucleic acid-binding proteins. For example, oxidation of the iron-responsive element binding protein (IRE-BP), which specifically interacts with the 5′ untranslated region of the ferritin heavy-chain mRNA, increased its binding affinity to its RNA target (IRE) by a factor of over 100 (31). On the other hand, in the case of the bacterial transcriptional regulatory protein OxyR, which regulates gene expression in response to oxidative stress, oxidation of a single essential cysteine changed its DNA-binding specificity and activated gene expression of a set of genes responsible for the resistance to oxidative environment (32). Furthermore, the DNA-binding activity of the Fox- Jun heterodimer has been shown to be modulated by the redox status of a single conserved cysteine residue in the DNA-binding domain of each of the two proteins (33). However, similar experiments with other transcription factors, namely CBP, Sp-1 and NF-1 ((46) and our unpublished results) did not show any effects of oxidation or reduction of sulfhydryls on their DNA binding activities. Thus, one of the physiological roles of the cellular redox regulation system might be that it serves as a novel modality in selective activation of a certain set of genes by modulating the activity of particular nucleic acid-binding proteins. Besides the TRX system, there are other cellular reducing systems such as the glutaredoxin (GSH) system (29,48). The TRX and GSH systems are considered to have specific, but overlapping functions in many reduction systems. Since reduced glutathione, even in the presence of glutathione reductase, did not restore the activity of the oxidized NFxB in vitro (T. Kawabe, T. Okamoto, and Y. Yodoi, submitted), TRX is more likely to be important in the reduction of NFxB. Recently, Staal et al. (49) reported that N-acetyl-L-cysteine (NAC), acting both as a scavenger for oxygen radicals and as a precursor of GSH, blocked activation of HIV transcription by TN F-α through NFxB when added at a very high concentration. It is possible that there are cross talks within the cells among different reducing systems. However, the in vivo effects of reducing agents, especially in the presence of oxidative stress, such as that evoked by TNF-α, need to be evaluated carefully. For example, the effect of TNF- α in inducing endogenous TRX may have been blocked in the presence of excess intracellular GSH because of the radical scavenger activity of GSH. Thus, cellular reducing systems, such as those of TRX and GSH, might have distinct roles in the signal transduction pathway involving NFxB. In T cells infected with HTLV-1, where TRX (or ADF) (25-27) as well as TNF (16) is overexpressed, NFxB is known to be constitutively activated (15,22) and replication of HIV is greatly augmented (23,24). It is interesting to see if excessive amounts of the extracellular reducing agent such as NAC could inactivate NFxB even in cells infected with HTLV-1.
There are many cysteine residues in the putative DNA-binding domain of the NFxB molecule, but the cysteine residue(s) required for DNA recognition has not yet been identified. Nevertheless, our results, together with others (34- 36), suggest that a novel molecular mechanism involving the sulfhydryls has an important role in specific DNA recognition by NFxB. The concept of ‘redox regulation’ may not only provide an insight into a novel mechanism of DNA-binding by NFxB but also may explain, at least partly, why gene expression of HIV and IL-2Rα is augmented in the cells transformed by HTLV-1. Further investigation will hopefully lead to development of a new strategy which can efficiently decelerate the pathological process of AIDS.
Acknowledgements
We thank Drs A. Holmgren, T. Tursz, T. Hirakawa, N. Arai, S. Kato, H. Masutani, and K. Shimotohno for helpful discussions. This work was supported by grants-in-aid from the Ministry of Health and Welfare and the Ministry of Education, Science and Culture of Japan.