| Title | Nitric oxide synthesis and TNF-α secretion in RAW 264.7 macrophages Mode of action of a fermented papaya preparation |
|---|---|
| Year | 2000 |
| Author | Gerald Rimbacha, Young Chul Parka, Qiong Guoa, Hadi Moinia, Nilofer Qureshib, Claude Salioua, Kuni Takayamab, Fabio VirgiliC, Lester Packer, |
| Publisher | Life Sciences. |
Nitric oxide synthesis and TNF-α secretion in RAW 264.7 macrophages
Mode of action of a fermented papaya preparation
Gerald Rimbacha, Young Chul Parka, Qiong Guoa, Hadi Moinia, Nilofer Qureshib, Claude Salioua, Kuni Takayamab, Fabio VirgiliC, Lester Packer·a,*
a Department of Molecular and Cell Biology, 251 Life Science Addition, University of California, Berkeley, CA 94720-3200, USA
bMycobacteriology Research Laboratory, William S. Middleton Memorial Veterans Hospital, Madison, WI, USA
c National Institute of Nutrition, Rome, Italy
Abstract
Macrophage inducible nitric oxide synthase is able to generate massive amounts of nitric oxide (NO) which contributes to the host immune defense against viruses and bacteria. Monocyte-macrophages stimulated with the bacterial wall component lipopolysaccharide (LPS) and cytokines such as interferon-γ (IFN-γ) express the inducible form of nitric oxide synthase (iNOS). Furthermore, tumor necrosis factor-α (TNF-α) is one of the central regulatory cytokines in macrophage antimicrobial activity and synergizes with IFN-γ in the induction of NO synthesis. Because of its pivotal role in both antimicrobial and tumoricidal activities of macrophages, a significant effort has focused on developing therapeutic agents that regulate NO production. In the present study fermented papaya preparation (FPP) is shown to exert both immunomodulatory and antioxidant activity in the macrophage cell line RAW 264.7. Interestingly, a low and a high molecular weight fraction (LMF and HMF, respectively) of FPP exhibited different activity patterns. FPP fractions alone did not affect NO production. However in the presence of IFN-γ, both LMF and HMF significantly increased iNOS activity and nitrite as well as nitrate accumulation. NO radical formation measured in real-time by electron paramagnetic resonance spectroscopy was higher in the presence of LMF and IFN- γ. On the contrary, iNOS mRNA levels were enhanced further with HMF than with LMF. Moreover, LMF displayed a stronger superoxide anion scavenging activity than HMF. In the presence of iFN-γ, both FPP fractions stimulated TNF-a secretion. However in non-stimulated macrophages, TNF-α secretion was enhanced by HMF only. Since water-soluble FPP fractions contained no lipid A, present data indicate that FPP is a macrophage activator which augments nitric oxide synthesis and TNF-α secretion independently of lipopolysaccharides. © 2000 Elsevier Science Inc. All rights reserved.
Keywords: Nitric oxide; Tumor necrosis factor-alpha; RAW 264.7 macrophages; Fermented papaya preparation; Bionormalizer™
* Corresponding author. Tel.: 510-642-1872; fax: 510-642-8313
E-mail address: packer@socrates.berkeley.edu (L. Packer)
0024-3205/00/$- see front matter © 2000 Elsevier Science Inc. All rights reserved.
PII: S0024-3205(00)00664-0
Introduction
Nitric oxide (NO) is a multiple regulatory molecule that is involved in a wide range of physiological and pathophysiological processes (1). NO is produced in mammalian cells constitutively or upon induction of a family of distinct enzymes known as nitric oxide synthase (NOS). Appropriate stimuli for the expression of the inducible form of nitric oxide synthase in monocyte-macrophages are the exposure to the bacterial wall components lipopolysaccharide (LPS) and various cytokines such as interferon-γ (IFN-γ, tumor necrosis factor-α (TNF-α) and interleukin-1β (2). Macrophage inducible nitric oxide synthase (iNOS) is able to generate massive amounts of NO which contributes to the host immune defense against viruses and bacteria. However, high NO production has been associated with oxidative stress and with the pathophysiology of various diseases such as arthritis, diabetes, stroke, septic shock, autoimmune diseases, and chronic inflammation (3). Because of its pivotal role in the antimicrobial and tumoricidal activities of macrophages, a significant effort has focused on developing therapeutic agents that regulate NO production or action (4,5). Furthermore, various plant extracts have been reported to protect from NO-induced oxidative stress (6-9).
Fermented papaya preparation (FPP) is made by yeast fermentation of Carica papaya Linn., Pennisetum purpeum Schum. and Sechium edule Swartz. FPP is used as a natural food health supplement also known as Bionormalizer in different parts of the world. FPP has been reported as an antioxidant able to scavenge hydroxyl radicals in vitro (10) and to protect the brain of aged rodents in vivo challenged either by oxidative stress (11), by iron treatment (12) or by ischemia-reperfusion injury (13). Furthermore, the accumulation of thiobarbituric acid reactive substances and protein carbonyls were found to be lower in heart homogenates from FPP supplemented rats exposed to peroxyl radicals as compared to non-supplemented controls (14,15). From these reports it has been proposed that beneficial effects of FPP might be due to its free radical scavenging properties.
FPP has also been shown to upregulate phorbol ester or zymosan-induced superoxide production in rat peritoneal macrophages (16), natural killer cell activity (17) and the level of IFN-γ in human blood (18). Such evidence suggests that FPP also possess the ability to modulate immune effector cells in addition to its direct free radical scavenging activity. Recent studies clearly demonstrated that FPP affects NO and hydrogen peroxide production in macrophages, respectively (19). However, little is known about the mode of action of FPP and which particular constituents of FPP are mediating its cellular activity. In the present study, the activity of either a low or a high molecular weight fraction of FPP was investigated with regard to their effect on NO synthesis, superoxide anion scavenging activity and TNF-α secretion in the mouse macrophage cell line RAW 264.7.
Materials and methods
Chemicals and materials
Murine recombinant IFN-γ (1X106 U/mg) and murine recombinant TNF-α (1X106 U/ml), rabbit anti-murine TNF-α polyclonal antibody (neutralizing), and hamster anti-murine TNF-α monoclonal antibody were purchased from Genzyme (Munchen, Germany). LPS (phenol extracted Salmanella enteritidis), sodium nitrite, sulfanilamide, N-(1-naphthyl)-ethylenediamine dihydrochloride, protease inhibitor cocktail, L-arginine, flavin-adenine dinucleotide, dithiothreitol (DTT), the reduced form of nicotinamide-adenine dinucleotide phosphate (NADPH), lactate dehydrogenase, p-nitrophenyl phosphate, xanthine oxidase, nitrate reductase and 5,5-dimethyl-1-pynoline-N-oxide (DMPO) were purchased from Sigma Chemical Co. (St. Louis, MO). The spin-trap reagent for trapping NO was the [(MGD)2-Fe2+] complex, prepared by reacting N-methyl-D-glucamine dithiocarbamate (MGD) (25 mM) with FeSO4 (5 mM). The FeSO4 solution was freshly prepared for each experiment. Fetal calf serum was obtained from University of California, San Francisco cell culture facility. RPMI containing L-arginine (200 mg/L), HBSS, and other tissue culture reagents were purchased from Life Technologies (Gaithersburg, MD). Silica gel GHL thin layer plates (250 µM) were obtained from Analtech Inc., Newark. FPP (Bionormalizer™) was provided by Osato Research Foundation, Japan.
Preparation of FPP fractions
FPP was suspended in Dulbecco’s PBS (pH 7.4) for 30 min. After centrifugation at 15,000 g, the supernatant containing the water-soluble FPP fractions was subjected to a molecular weight cut filter (Centricon, C-3, MW cut off = 3,000) and also centrifuged for 3 h at 7500 g in order to obtain a low molecular weight fraction (LMF) and a high molecular weight fraction (HMF). Each fraction was diluted in Dulbecco’s PBS in order to equate it to the original concentration of FPP.
Macrophage culture
The murine monocyte/macrophage cell line RAW 264.7 was obtained from the American Type Culture Collection (Rockville, MD). Cells were maintained in 75 cm2 plastic flasks (Falcon, NJ) with RPMI 1640 supplemented with 10% fetal calf serum and antibiotics. For experiments, macrophages were detached by vigorous pipetting and, after centrifugation, incubated with fresh medium in either 96-well tissue culture plates (2X105 cells/well) or 100 mm diameter plastic petri dishes (1X107 cells/dish) for 3 h at 37°C in an atmosphere of 5% CO2 plus air. Then, these cells were treated with IFN-γ and/or FPP and cultured for 24 h.
Nitrite and nitrate accumulation
Experiments were performed on cells grown either under standard conditions or in the presence of LMF and HMF with or without IFN-γ for 24 h under the conditions described in the figure legends. Supernatants in cultured macrophages were collected and mixed with an equal volume of the Griess reagent (1% sulfanilamide I 0.1% N-(1-naphthyl)-ethylenediamine dihydrochloride / 2.5% H3PO4) and incubated for 10 min at room temperature. Nitrite (NO2–) concentration was determined by measuring the absorbance at 540 nm in a Titertek Multiskan (Flow Laboratories, North Ryde, Australia). Cell-free medium alone contained 5 µM of NO2–; this value was determined in each experiment and subtracted from the value obtained with cells. Nitrate (NO3–) was measured by reducing nitrate to nitrite with bacterial nitrate reductase, then measuring nitrite by using the Griess reagent (20).
iNOS activity
Macrophage monolayers were washed three times with PBS, scraped into PBS, and centrifuged at 500 g for 15 min at 4°C. The cell pellet was resuspended in 500 µ-1 of sonication buffer, which contained 40 mM Tris-HCl buffer (pH 8) and a protease inhibitor cocktail and then lysed by sonication. Aliquots of the lysate were used for Bradford protein assay (BioRad, Richmond, CA). iNOS enzyme activity was measured as described (21). Briefly, 50 µg of macrophage lysate were incubated for 2 h at 37°C in 20 mM Tris-HCl (pH 7.9) containing 4 µM BH4, 4 µM FAD, 3 mM DTT, and 2 mM each L-arginine and NADPH. The reaction was initiated by adding L-arginine and NADPH and stopped by adding lactate dehydrogenase in order to oxidize residual NADPH (22). Nitrite and nitrate were measured by the Griess reaction.
Reverse transcription polymerase chain reaction
Total RNA was extracted from cells following the method of Chomczynki and Sacchi (23). Rerverse transcription was performed using a RNA PCR kit (Perkin-Elmer, Branchburg, NJ). One microgram of total RNA was reverse transcript to cDNA following the manufacturer’s recommended procedures. Reverse transcriptase generated cDNA encoding iNOS and G3PDH (Glycerinaldehyde 3-phosphate dehydrogenase, as a positive control and internal standard) genes were amplified using polymerase chain reaction (35 cycles). Oligonucleotide primers that correspond to the mouse macrophage iNOS and murine G3PDH cDNA were purchased from Clontech and used following the manufacturer’s procedures. PCR was also performed using a RNA PCR kit. The reaction volume was 40 µl containing (final concentration): PCR buffer (1X) deoxynucleotide (0.2 mM each), MgCl2 (2 mM), Taq DNA polymerase (2 U), oligonucleotide primer (0.5 M each), and reverse transcriptase products. After initial denaturation for 2 min at 95°C, 35 cycles of amplification (95°C for 1 min, 65°C for 1 min, and 72°C for 1.5 min) were performed followed by a 7 min extension at 72°C. A 10 µl aliquot from each PCR reaction electrophoresed in a 1.7% agarose gel containing 0.2 µ-g/ml ethidium bromide. The gel was then photographed under ultraviolet light.
EPR measurements
Besides use of the Griess colorimetric assay as a means for the indirect measurement of NO production, now more direct and sensitive methods for detection of NO are available. Since NO is paramagnetic and reacts to form high-affinity nitroso complexes with a variety of metal complexes, the distinctive electron paramagnetic resonance (EPR) spectra of these nitroso complexes permit quantitative measurements of NO generation in biological systems (24). In comparison to the indirect measurement of NO production EPR spin trapping provides a sensitive, direct and continuous technique allowing a real-time detection of the formation and decay of NO radical near to the site of generation. EPR spectra were recorded by using an IBM ER 200D-SRC EPR spectrometer, as follows: central field 3420 G; modulation frequency 100KHz; modulation amplitude 3.2 G; microwave power 20 mW; scan width 200 G, gain 6.3X105; temperature 25°C. Macrophages were plated at 2Xl07 cells/100 mm and were cultured for 16 h in either medium alone or medium containing IFN-γ (50 U/ml) or IFN- γ plus FPP (LMF or HMF, 6 mg/ml). After adding the spin-trap complex [(MGD)2–Fe2+] and L-arginine (0.7 mM), cells were incubated for 90 min at 37°C and then transferred to the chamber for the measurement of NO.
Superoxide radicals (O2˙- ) were generated by the hypoxanthine-xanthine oxidase system and the EPR spectra of the DMPO-OOH spin adducts were recorded (25). Conditions were as above except the central field was 3475 G, the modulation amplitude 2.0 G, and the scan width 100 G. The percentage superoxide anion radical scavenging effect (E) of FFP fractions was calculated as follows: E = (h0 -hx/h0) X 100%; here h0 and hx were the ESR signal intensities of samples without and with FFP fractions, respectively.
Xanthine oxidase activity
Xanthine oxidase activity was determined spectrophotometrically by following the production of uric acid at 295 nm (Σ= 11 M-1cm-1). Reaction mixture contained 150 µM xanthine and 0.1 mM EDTA in 50 mM potassium phosphate buffer (pH = 7.4). Milk xanthine oxidase activity was measured in the presence or absence of either FPP, or its molecular weight fractions HMF and LMF.
Electrophoretic mobility shift assay
One hour after treatment the nuclear extracts were prepared according to the method described by Suzuki and Packer (26). Nuclear extracts (10 µg) were preincubated with 2 µg poly (dl-dC) at 2°C and then incubated with 32P-labeled KB probes for 20 min at room temperature before separation by electrophoresis through 6% polyacrylamide gel and subsequent autoradiography (27).
Assay ofTNF-α secretion
TNF-α secretion was measured by modification of an enzyme linked immunosorbent assay (ELISA), as described earlier (28). The sensitivity of the ELISA to TNF-α concentrations in medium was above 40 pg/ml. The ELISA was devised by coating 96-well plates with 6.25 ng/well of murine monoclonal antibody with specificity for murine TNF-α. Before use and between subsequent steps in the assay, coated plates were washed twice with PBS containing 0.05% Tween-20 and twice with PBS alone. All reagents used in this assay were incubated for 1 h at room temperature with coated wells.
For the standard curve rTNF-α was added to serum previously determined to be negative for endogenous TNF-α. After exposure to the medium, assay plates were sequentially exposed to rabbit anti-TNF-α, phosphatase-conjugated goat anti-rabbit IgG, and p-nitrophenyl phosphate. Optical density readings at 410 nm were taken using a Emax 96-well microtest plate spectrophotometer (Molecular Devices, Menlo Park, CA).
Limulus amoebocyte lysate assay
LMF and HMF were tested for LPS and LPS-like compounds using the Limulus amoebocyte lysate assay (Sigma Chemical Co., St. Louis, MO). Briefly, when exposed to minute quantities of LPS or LPS like compound from the walls of gram-negative bacteria, the lysate increases in opacity as well as in viscosity thereby forming a gel depending on the concentration of endotoxin.
Lipid A analysis
FPP, HMF and the water-insoluble pellet which remains after the centrifugation step for the preparation of the water-soluble FPP fractions were tested for the presence of lipid A. Samples (FPP and HMF) and reference standard (deep rough chemotype lipopolysaccharide from Eschericha coli D31m4) were treated with EDTA, and purified by a DEAE cellulose column chromatography as previously described (29).
Samples were hydrolyzed in 2 ml of 0.1 M HCl at 100°C for 30 min to yield monophosphoryl lipid A. Then 5 ml of chloroform/methanol (2:1, v/v) was added, mixed and centrifuged. The lower organic layer was recovered, filtered and dried. The extracted samples were applied in chloroform/methanol (4:1, v/v) to silica gel GHL plate (250 µm) and irrigated with chloroform /methanol /water / concentrated ammonium hydroxide (50:25:4:2, v/v). Spots were visualized by spraying the plate with dichromate/sulfuric acid reagent and charring.
Statistical analysis
Data are expressed as mean values ± standard deviation (SD). One-way analysis of variance (ANOVA) followed by the Scheffe multiple range test was used for statistical analysis (Statistical Package for the Social Sciences, SPSS for Windows, Vers. 6.0.1). Differences were considered significant at the p < 0.05 level. Data in tables and figures are presented either as the mean of at least three distinct experiments (± SD) or a representative experiment was shown.
Results
Effect of FPP fractions on nitric oxide synthesis in IFN-γ-stimulated macrophages
NO generated by NOS, spontaneously oxidizes forming both NO2– and NO3–. Moreover, macrophages stimulated by LPS and IFN- γ also activate the enzymatic machinery producing O2˙–, which in turn reacts with NO generating peroxynitrite (30). Decomposition of peroxynitrite leads to a recombination of radicals decaying largely to NO3– but also to NO2–. Non activated macrophages produced low levels of NO (< 5 µM) constitutively. Treatment with IFN-γ significantly increased the production of NO2– and NO3– (Table 1). When IFN-γ stimulated macrophages were simultaneously treated with FPP (3 mg/ml) a 3 fold greater increase of NO synthesis was observed in comparison with untreated cells. Both the low and high molecular weight fraction of FPP significantly enhanced IFN-γ induced NO production in the macrophage cell line RAW 264.7. The upregulation of IFN-γ induced NO production by FPP and its fractions was already significant at 0.25 mg/ml, and reached a plateau levels at 3 mg/ml. FPP alone or in combination with IFN-γ was not toxic for macrophages, as assessed by trypan exclusion.
Table 1
Effects of FPP and its low (LMF) and high molecular weight fraction (HMF) on nitrite and nitrate synthesis in RAW 264.7 cells. Macrophages (2X105) were treated in the absence and presence of IFN-γ (10 U/ml) with 3 mg/ml FPP, LMF or HMF for 24 hours
| Treatment | NO2- | NO3- |
|---|---|---|
| None | 22 ± 3a | 19 ± 3a |
| FPP | 63 ± 7c | 78 ± 7b |
| LMF | 47 ± 5b | 79 ± 6b |
| HMF | 58 ± 6bc | 80 ± 6b |
Values for nitrite plus nitrate synthesis are given in brackets. Values within a column with different superscripts are significantly different (P < 0.05).
FPP fractions and iNOS activity in macrophages
Different plant extracts have been reported to affect the activity of various enzymes either in vitro and in other cellular models. In particular, both the constitutive and the inducible NOS activities have been shown to be affected by plant derived complex mixtures of polyphenols and bioflavonoids. It therefore was of interest whether FPP and fractions derived from FPP were able to affect the activity of the inducible form of the nitric oxide synthase activity. These experiments sought to characterize the mechanism underlying the upregulation in IFN-γ induced NO synthesis by LMF or HMF. Table 2 shows that IFN-γ alone significantly increased iNOS enzyme activity. In the presence of IFN-γ both LMF and HMF resulted in a further 4-5 fold increase in the activity of iNOS. However in the absence of IFN-γ neither LMF nor HMF affected iNOS enzyme activity as compared to untreated control macrophages.
Table 2
Effects of the low (LMF) and high molecular weight fractions (HMF) of a fermented papaya preparation on iNOS enzyme activity in RAW 264.7 cells (conditions as in Table 1)
|
Treatment |
NO synthase activity |
|
|
FPP fraction |
IFN- γ |
|
|
– – LMF LMF HMF HMF |
– + – + – + |
13 ± 3a 67 ± 5b 14 ± 5a 257 ± 31c 14 ± 3a 315 ±40c |
Values within a column with different superscripts are significantly different (P < 0.05).
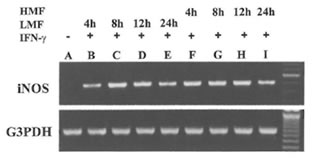
Fig.1. Induction of the expression of iNOS mRNA in RAW 264.7 cells by LMF or HMF. Macrophages (2 X 105) were treated with IFN-γ (10 U/ml) plus LMF (3 mg/ml) or HMF (3 mg/ml) for various time points as indicated. Lane A: Control (no treatment), Lane B- E: 4, 8, 12, and 24 h treatment with LMF and IFN-γ; Lane F- I: 4, 8, 12 and 24 h treatment with HMF and IFN-γ.
Effect of FPP fractions and iNOS mRNA expression in macrophages
Besides the effect on iNOS enzyme activity, the effect of treatment with LMF or HMF on the expression of the iN OS gene at the transcriptional level was investigated. RAW 264.7 macrophages did not express iNOS mRNA levels constitutively. As shown in Fig. 1, the combination of IFN-γ with LMF was associated with an increase of iNOS mRNA expression with a peak after 8 and 12 h of supplementation, respectively. In comparison to LMF, HMF treatment caused a more prolonged increase of IFN-γ-induced iNOS mRNA expression that was already observed after 4 h supplementation. After 24 h of LMF or HMF treatment lower iNOS mRNA levels were evident as compared to the corresponding earlier timepoints (8 and 12 h). These results demonstrate that in the presence of IFN-γ, both LMF or HMF modulate iNOS mRNA expression, thereby apparently exhibiting different kinetics.
FPP fractions and real-time NO radical formation in macrophages
In order to measure real-time NO radical production by RAW 264.7 macrophages an EPR spin-trap technique was applied. In this system NO was trapped by a complex consisting of
MGD and reduced iron to form a stable and water soluble [(MGD)2-Fe2+-NO] adduct. In non-activated macrophages or after treatment of macrophages with LMF or HMF alone no EPR signal was detected. However, a typical three-line EPR spin trapped NO signal was observed when macrophages were stimulated with IFN-γ (inset of Fig. 2).Its hyperfine splitting constant (aN = 12.5 G) is the same as reported in the literature (31). The simultaneous addition of IFN-γ plus LMF as well IFN-γ plus HMF resulted in an increase of the corresponding EPR signal intensity of the [(MGD)2-Fe2+-NO] spin adduct. As shown in Fig. 2, after 8, 12, and 16 h treatment, LMF had a more profound effect on the real-time NO radical formation than HMF.
Superoxide radical scavenging activity and effect on xanthine oxidase activity of FFP fractions in vitro
The typical EPR spectrum of spin adduct DMPO-OOH was detected (the inset in Fig. 3, aN = 14.6 G, aHβ = 11.4 G, aH γ = 1.2 G) in the hypoxanthine-xanthine oxidase system using the spin trapping agent DMPO. The superoxide radical-scavenging activity of the two fractions of FPP was dose-dependent (Fig. 3). Concentrations of LMF or HMF scavenging 50% of the superoxide anion radical (IC50) were 7 and 20 mg/ml, respectively. FPP, LMF and HMF, up to a concentration of 30 mg/ml did not affect xanthine oxidase activity in vitro (data not shown).
FPP fractions and TNF-a secretion in macrophages
TNF- α is one of the central regulatory cytokines in the induction of macrophage antimicrobial activities (32). Furthermore TNF-α synergizes with IFN-γ known to induce NO synthesis (33). Therefore it was of particular interest whether FPP and its low and high molecular weight fractions affect TNF-α secretion in the macrophage cell line RAW 264.7. As shown in Fig. 4, non-activated macrophages secreted negligible amounts of TNF-α into the medium. However, IFN-γ substantially enhanced the production of TNF-α. Simultaneous treatment of IFN-γ with FPP as well as its low and high molecular weight fraction resulted in a further increase in TNF- α secretion. In the absence of IFN-γ, TNF- α secretion was significantly enhanced by FPP and HMF whereas treatment with LMF alone resulted in no increase in TNF-α secretion.
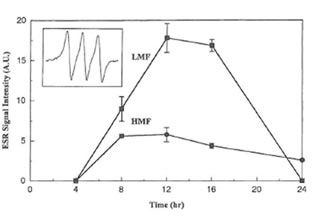
Fig. 2. Time course of EPR signal intensities of the [(MGD)2–Fe2+-NO] spin adduct produced in RAW 264.7 cells stimulated by IFN- γ (50 U/ml) plus LMF (6 mg/ml) or HMF (6 mg/ml) for various time periods. Inset shows the hyperfine splitting of the [(MGD)2–Fe2+-NO] spin adduct.
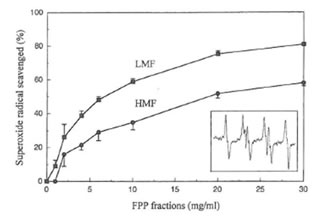
Fig. 3. Superoxide radical scavenging activity of LMF or HMF. Inset shows the hyperfine splitting of the DMPO-OOH spin adduct.
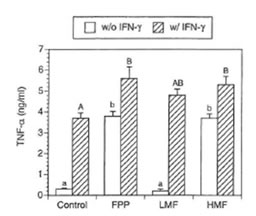
Fig. 4. Effects of FPP and its low and high molecular weight fraction on secretion of TNF- α by RAW 264.7 cells (conditions as in Table 1). Bars with different small (without IFN-γ treatment) and capital letters (with IFN-γ treatment) are significantly different (P – 0.05).
Effect of FPP fractions on NF-KB activation in macrophages
It is now accepted that the redox status of cell may have an effect on certain transcription factors and regulate gene expression (34). In macrophages, the synthesis of the inducible form of NO synthase induced by IFN-γ is dependent on the activation of a specific kinases and on the subsequent transfer to the nucleus and DNA binding by the transcription factor NF-KB. In agreement with these data, the expression of iNOS has been reported to be induced by oxidative stress and the resulting production of NO inhibits NF-KB binding to DNA thus acting as a feed-back regulator of the overall NO flow through the cell (35). Therefore we sought to investigate whether the observed changes of NO production by LMF or HMF was due to any effect at the transcriptional level. As shown in Fig. 5 treatment with only FPP fractions was not associated with a significant increase of NF-KB binding in comparison with untreated control cells. However in the presence of IFN-γ the DNA binding activity of NF-KB was affected by both LMF and HMF, suggesting that the regulation of iNOS expression by FPP fractions in the presence of IFN-γ is at least in part due to an enhancement of the signal at the transcriptional level.
Limulus amoebocyte lysate assay and lipid A analysis
HMF showed a positive Limulus amoebocyte lysate test indicated by the formation of a hard gel which permitted a complete inversion of the tube without disruption of the gel. LMF showed a negative Limulus amoebocyte lysate test. Based on monophorsphoryl lipid A analysis FPP and its high molecular weight fraction were completely devoid of LPS. However, the pellet fraction showed the presence of barely dectectable amounts of LPS as shown in Fig. 6.
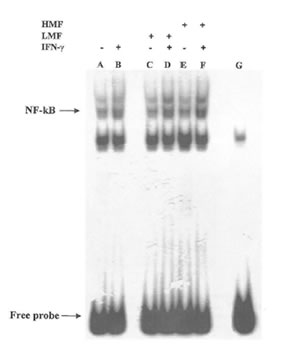
Fig. 5. Effects of LMF or HMF on NF-KB DNA binding in IFN-γ stimulated macrophages. Macrophages (2X105) were stimulated with different concentrations of LMF or HMF in the presence or absence of IFN-γ (10U/ml) and incubated for 2 h. Activation of NF-KB was assayed using electrophoretic mobility shift assay. Lane A: control (no treatment); lane B: IFN-γ (10 U/ml); lane C: LMF (3 mg/ml); lane D: IFN-γ plus LMF; lane E: HMF (3 mg/ml); lane F: IFN-γ plus HMF; lane G: cold probe competition.
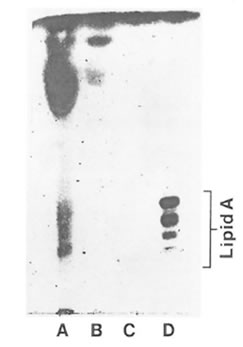
Fig. 6. Analytical silica gel thin layer chromatography of acid-hydrozyed FPP fractions. Lane A: water insoluble pellet fraction; lane B: FPP (entire fraction); lane C: HMF; lane D: ReLPS purified from Eschericha coli. The standard lipid A is shown to consist of a series of bands ranging from hexaacyl-. pentaacyl-, tetraacyl-, and triacyl monophosphoryl lipid A (top to bottom).
Discussion
It has been demonstrated that FPP affects NO synthesis, and TNF-α secretion in RAW 264.7 macrophages. Furthermore FPP displays antioxidant activity in terms of its superoxide anion radicals. These properties of FPP may be, in part, responsible for its reported beneficial activities toward various diseases. Interestingly, a low and high molecular weight fraction of FPP exhibited different mechanism and kinetics modulating the antioxidant / oxidant balance as well as the secretion of the proinflammatory cytokine TNF-α in macrophages.
FPP fra ctions trigger NO accumulation and change iNOS gene expression and iNOS activity
FPP has been reported to induce natural killer cell activity (17) and to enhance the capacity of respiratory burst in neutrophils in vitro (16) supporting the hypothesis that FPP acts as an immune modulator. Therefore the underlying mechanisms by which FPP affects NO production and TNF-α secretion in RAW 264.7 macrophages are of interest. Treatment of macrophages with LMF or HMF alone failed to induce appreciable levels of NO production. However, significant increases of NO production occurred after cells were treated with a combination of IFN-γ with either LMF or HMF. These results are in agreement with our previous studies which utilized non-fractionated FPP (19). Hence FPP itself does not provide a priming signal able to induce the NO pathway. Since FPP acts to enhance NO production in the presence of IFN-γ it was of interest to determine how it regulates NO metabolism. It is known that iNOS activity is regulated mainly at transcriptional and post-transcriptional level at different regulatory steps (2). To characterize the mechanism by which FPP changes IFN-γ induced NO production in macrophages, the effects of LMF or HMF on iNOS enzyme activity, iNOS mRNA expression, and real-time NO radical formation were tested. In the presence of IFN-γ, LMF significantly enhanced iNOS enzyme activity indicating that this fraction contains compounds which can act as modulators of the iNOS. Treating cells with increasing amounts of glucose up to 25 mM (data not shown) also resulted in enhanced nitrite accumulation and iNOS activity. This observation possibly supports the hypothesis that LMF (analysed final glucose concentration in 3 mg/ml LMF = 15 mM) at least partially mediates the enhancing effect on NO production due to a glucose-dependent effect. In the case of iNOS mRNA levels it is know that the exposure of macrophages to high glucose for 24 h leads to a stimulation of iNOS gene expression (36). A potential mechanism of the high glucose-enhancing effect on iNOS mRNA may be due to protein kinase C activation. High glucose has been shown to increase the de novo production of diacylglycerol which is known as a potent activator of protein kinase C in many cell types (37). Another signaling molecule of potential interest regarding LMF is cyclic AMP. High glucose has been reported to increase cyclic AMP in murine mesangial cells; thus has been implicated in mediating iNOS gene expression thus raising the possibility that the observed effect of LMF on iNOS mRNA may also be mediated via increased levels of cyclic AMP (38).
Expression of the iNOS gene is regulated by endotoxin and cytokines such as IFN-γ and interleukin-2. Two upstream DNA regions of the iNOS promoter, the RI and RII domains, are required for the maximal promoter activation by LPS, and the RII domain mediates promoter trans-activation of IFN-γ. Both of these domains comprise multiple sequences homologous to those of cis elements involved in transcription activation, such as NF-KB binding sites, IFN-γ response elements, and IFN-γ-activated factor binding sequence (39). LMF and HMF affect NF-KB DNA binding which account for the observed augmentation of the iNOS gene transcription. We previously reported that the halflife of iNOS mRNA in macrophages treated with IFN-γ plus FPP was approximately 7 h and it was not significantly different from that cells treated with IFN-γ alone (19). This suggests that the mechanism by which FPP augments iNOS mRNA expression is not due to an increase in the stability of iNOS mRNA. Hence it is likely that FPP modulates IFN-γ-induced induction of iNOS by upregulating the rate of gene transcription by as yet unknown mechanism. Furthermore, after 24 h treatment with either LMF or HMF, a significant decrease in iNOS mRNA levels in the presence of IFN-γ was evident. This may occur because NO exerts a negative feed-back mechanism on its own production which may be important for limiting excessive or prolonged NO synthesis as previously described in J744 macrophages (35).
Modulation of NO radical formation by FPP and antioxidant activity
NO was trapped by the complex consisting of MGD and reduced iron (Fe2+) to form a stable and water soluble [(MGD)2-Fe2+-NO] complex. As reported elsewhere (40), no EPR signal was detected in both non-stimulated macrophages or macrophages treated only with LMF or HMF. However, when IFN-γ was present a substantial increase in real-time NO radical formation after LMF or HMF treatment was observed. In contrast to the data obtained for nitrite accumulation, higher EPR signal amplitudes in LMF as compared to HMF treated macrophages were evident. It is known that the simultaneous production of superoxide and NO results in the rapid formation of peroxynitrite by macrophages (41). Under the conditions investigated addition of superoxide dismutase to the stimulated macrophages resulted in a marked, concentration- dependent increase in the production of NO radicals (data not shown) as described previously (42). Furthermore as in the present study and in other investigations (10), it has been found that FPP scavenges superoxide anion radicals generated by the hypoxanthine-xanthine oxidase system. Therefore, differences in NO radical formation between LMF and HMF might be directly related to differences in the superoxide anion scavenging activity of the two FPP fractions. Since LMF had a much higher superoxide anion radical scavenging activity than HMF, a smaller quantity of NO radicals were possibly converted into peroxynitrite which finally leads into a higher NO radical formation in LMF-treated cells. Moreover it should be also taken into consideration that the activity of xanthine oxidase used as the radical generating system for the EPR studies remained unchanged even in the presence of very high concentrations of LMF or HMF (up to 30 mg/ml). Thus FPP contains compounds, not identified yet, which can directly function as antioxidants in terms of superoxide anion radical scavenging. The present data also clearly demonstrate that nitrite accumulation as an index of NO production cannot directly reflect real-time NO production in macrophages as has already been pointed out elsewhere (31).
Effect of FPP fractions on TNF-α secretion
It is well documented that induction of iNOS gene expression is regulated tightly by mediators such as interleukin-2, IFN-γ, and inflammatory stimuli such as bacterial LPS. Furthermore it is known that the production of TNF-α is crucial for the synergistic induction of NO synthesis in IFN-γ and/or LPS-stimulated macrophages (43). TNF-α elicits a number of physiological effects including septic shock, inflammation, cachexia and cytotoxicity (44). These effects depend upon the interaction of TNF-α with specific TNF-a receptors on cell surfaces (45). It has been shown in our previous study that in the presence of IFN-γ, FPP caused a manifold increase in TNF-a mRNA expression (19). However, little is known whether FPP also affects TNF-a at the protein level. Therefore this was investigated and it was found that both LMF and HMF resulted in a significant increase in TNF-a secretion when IFN-γ was present. This elevated TNF-a secretion due to FPP might finally result in a further augmentation of iNOS synthesis thereby possibly potentiating the microbicidal and tumoricidal activity of macrophages. However, in the absence of IFN-γ, HMF but not LMF increased TNF-a secretion into the medium. Thus the two fractions of FPP modulate TNF-a secretion in a different way. In this regard it has been recently demonstrated that glucose (which is present in high amount in LMF) as well as advanced glycosylation end products enhance production of interleukin-6 and TNF-a in human peripheral blood monocytes in vitro when IFN-γ was present (46). In the absence of IFN-γ, HMF mediated TNF-a production was most probably regulated on the post transcriptional level since HMF alone did not affect NF-KB DNA binding known to be important for TNF-a gene expression.
In the present investigation, HMF showed a positive Limulus amoebocyte lysate test which is used to indicate the presence of minute quantities of endotoxins such as LPS known as a potent inducer of TNF- α synthesis. LPS as a major consitutent of the wall of gram-negative bacteria is composed of a lipid A moiety attached to a polysaccharide chain (47). When LPS is treated under mild acid conditions, free lipid A is liberated which can be detected by thin layer chromatography (48). Based on this analysis there was no evidence for the presence of lipid A in HMF. This finding strongly suggests that FPP contains no significant amounts of gram-negative bacterial type LPS to attribute to its biological activities. Furthermore, regarding nitrite and nitrate accumulation, HMF displayed activities different than LPS since LPS alone is known to induce NO synthesis in macrophages. It has to be taken into consideration that the conventional Limulus amoebocyte lysate test is not specific to LPS because of the presence of a factor G mediated coagulation pathway in the amoebocyte lysate which is triggered by (1-3)-beta-D-glucans (49). Thus beside LPS also (1-3)-beta-D-glucans indicate positive results. Moreover, beta-D-glucans with a high molecular weight have been recently reported to stimulate TNF- α synthesis in RAW 264.7 macrophages. It is hypothesized that macrophages may incorporate beta-glucans through a beta-D-glucan specific mechanisms and/or endocytosis, which is finally associated with an enhanced cytokine production (50). Interestingly, in the presence of IFN-γ (1-3)-beta-glucans induced nitric oxide synthesis in macrophages (51). Similar to the present data with FPP, this combined effect of beta-D-glucans and IFN-γ was based on a priming effect of IFN-γ, because prestimulation with IFN-γ followed by beta-D-glucan treatment induced high nitric oxide production in macrophages, but reversal of this sequence of treatments had only a slight effect. Since FPP is made by yeast fermentation and (1-3)-beta-D-glucans are major structural constituents of the yeast cell wall, it is possible that some of the immunomodulatory effects of HMF reported here are directly related to (1-3)-beta-D-glucans which has to be clarified in further investigations.
Acknowledgments
Gerald Rimbach was supported by a grant (AZ Ri 884/3-1) from the German Research Society (DFG). Young Chul Park and Gerald Rimbach contributed equally to the work.
References
1. J.S. BECKMAN and W.H. KOPPENOL, Am J Physiol 271 Cl424-37 (1996).
2. C. NATHAN and Q.W. XIE, J Biol Chem 269 I 3725-8 (1994).
3. C. NATHAN, J Clin Invest 100 241 7-23 (1997).
4. R. REITER, L. TANG, J.J. GARCIA and A. MUNOZ-HOYOS, Life Sci 60 2255- 71 (1997).
5. R.J. REITER, Prog Neurobiol56 359- 84 (1998).
6. H. KOBUCHI, M.T. DROY-LEFAIX, Y. CHRISTEN and L. PACKER, Biochem Pharmacal 53 897- 903 (1997).
7. F. VIRGILI, H. KOBUCHI and L. PACKER, Free Radic Biol Med 24 1120- 9 (1998).
8. A.S. PANNALA, C.A. RICE-EVANS, B. HALLIWELL and S. SINGH, Biochem Biophys Res Commun 232 164-8 (1997).
9. J.E. CHESHIER, S. ARDESTANI-KABOUDANIAN, B. LIANG, M. ARAGHINIKNAM, S. CHUNG, L. LANE, A. CASTRO and R.R. WATSON, Life Sci 58 87-96 ( 1996).
10. L.A. SANTIAGO, J.A. OSATO, M. HIRAMATSU, R. EDAMATSU and A. MORI, Free Radic Bioi Med 11 379- 83 (1991).
11. L.A. SANTIAGO, J.A. OSATO, J. LIU and A. MORI, Neurochem Res 18 711-7 (1993).
12. L.A. SANTIAGO, J.A. OSATO, H. KABUTO and A. MORI, Med Sci Res 21139-141 (1992).
13. L.A. SANTIAGO. J.A. OSATO, N. OGAWA and A. MORI, Neuroreport 4 1031-4 (1993).
14. N. HARAMAKI, L. MARCOCCI, R. D’ANNA, L.J. YAN, H. KOBUCHI and L. PACKER, Biochem Mol Biol Int 36 1263-8 (1995).
15. L. MARCOCCI, R. D’ANNA, L.J. YAN. N. HARAMAKI and L. PACKER, Biochem Mol Biol Int 38 535-41 (1996).
16. J.A. OSATO, L.G. KORKINA, L.A. SANTIAGO and I.B. AFANAS’EV, Nutrition 11568-2 (1995).
17. D. OKUDA, H. OMIAMI, A. ZHOU, A. OSATO and L.A. SANTIAGO, Clin Rep 27 4249-8 (1993).
18. L.A. SANTIAGO, K. UNO, T. KISHIDA, F. MIYAGAWA, J.A. OSATO and L.A. SANTIAGO, Neurosciences 20 149-52 (1994).
19. H. KOBUCHI and L. PACKER, Biochem Mol Biol Int 43 14 1-52 (1997).
20. L.C. GREEN, D.A. WAGNER. J. GLOGOWSKI. P.L. SKIPPER, J.S. WISHNOK and S.R. TANNENBAUM. Anal Biochem 126 131-8 (1982).
21. Y. VODOVOTZ, N.S. KWON, M. POSPISCHIL, J. MANNING, J. PAIK and C. NATHAN, J Immunol 15241 10-8 (1994).
22. D.J. STUEHR. H.J. CHO, N.S. KWON, M.F. WEISE and C.F. NATHAN, Proc Natl Acad Sci U S A 88 7773-7 (1991 ).
23. P. CHOMCZYNSKI and N. SACCHI, Anal Biochem 162 156-9 (1987).
24. A. KOMAROY, D. MATTSON, M.M. JONES, P.K. SINGH and C.S. LAI, Biochem Biophys Res Commun 195 1191 (1993).
25. Y. NODA, K. ANZAI. A. MORI, M. KOHNO, M. SHINMEI and L. PACKER, Biochem Mol Bioi Intern 42 35-44 ( 1997).
26. Y.J. SUZUKI and L. PACKER, Biochem Biophys Res Commun 193 277-83 (1 993).
27. M.M. GARNER and A. REYZIN, Nucleic Acids Res 9 3047-60 (1981).
28. P. SCUDERI. K.E. STERLING, K.S. LAM, P.R. FINLEY, K.J. RYAN, C.G. RAY, E. PETERSEN, D.J. SLYMEN and S.E. SALMON, Lancet 2 1364-5 (1986).
29. N. QURESHI, K. TAKAYAMA, P. MASCAGNI, J. HONOVICH, R. WONG and R.J. COTTER, J Biol Chem 263 I 197 1- 6 (1988).
30. M.C. CARRERAS. J.J. PODEROSO. E. CADENAS and A. BOVERIS, Methods Enzymol 269 65- 75 (1996).
31. S.W. NORBY. J.A. WEYHENMEYER and R.B. CLARKSON, Free Radic Biol Med 22 1-9 (1997).
32. Y. WANG, D.S. HUANG and R.R. WATSON, Life Sci 54 401 – 11 (1994).
33. B.B. AGGARWAL. Biotherapy 3 113-20 (1991).
34. C. SALIOU. M. KITAZAWA, L. MCLAUGHLIN. J.P. YANG, J.K. LODGE, T. TETSUKA, K. IWASAKI, J. CILLARD. T. OKAMOTO and L. PACKER, Free Radic Biol Med 26 174-83 (1999).
35. F. D’ACQUISTO, L. SAUTEBIN. T. IUVONE. M. DIROSA and R. CARNUCCIO, FEBS Lett 440 76-80 (1998).
36. K. SHARMA. T.M. DAN OFF, A. DEPIERO and F.N. ZIYADEH, Biochem Biophys Res Commun 207 80-8 (1995).
37. P. FUMO. G.S. KUNCIO and F.N. ZIYADEH, Am J Physiol 267 F632-8 (1994).
38. D. KUNZ, H. MUHL, G. WALKER and J. PFEILSCHIFTER, Proc Natl Acad Sci USA 91 5387- 91 (1994).
39. A. MIZUTANI, H. MAKI, Y. TORII, K. HITOMI and N. TSUKAGOSHI. Nitric Oxide 2 235-41 (1998).
40. Y. XIA, V.L. DAWSON. T.M. DAWSON, S.H. SNYDER and J.L. ZWEIER, Proc Natl Acad Sci US A 93 6770-4 ( 1996).
4 1. W.A. PRYOR and G.L. SQUADRITO, Am J Physiol 268 L699- 722 (1995).
42. A.J. HOBBS, J.M. FUKUTO and L.J. IGNARRO, Proc Nall Acad Sci USA 91 10992-6 (1994).
43. C.D. JUN. S.M. CHOI. H.M. KIM and H.T. CHUNG, J Immunol l54 6541- 7 (1995).
44. B.B. AGGARWAL and K. NATARAJAN, Eur Cytokine Netw 7 93- 124 (1996).
45. S.I. LIOCHEV and I. FRIDOVICH, Free Radic Biol Med 23 668- 71 ( 1997).
46. M. MOROHOSHL K. FUJISAWA. I. UCHIMURA and F. NUMANO. Ann NY Acad Sci 748 562-70 (1995).
47. C.R. RAETZ, R.J. ULEYITCH, S.D. WRIGHT. C.H. SIBLEY, A. DING and C.F. NATHAN, Faseb J 5 2652-60 (1991).
48. K. TAKAYAMA. R.J. ROTHENBERG and A.G. BARBOUR, Infect Immun 55 231 1- 3 (1987).
49. T. OBAYASHI. H. TAMURA, S. TANAKA, M. OHKI. S. TAKAHASHI, M. ARAI, M. MASUDA and T. KAWAI. Clin Chim Acta 149 55-65 (1985).
50. M. OKAZAKI. Y. ADACHI, N. OHNO and T. YADOMAE, Biol Pharm Bull 18 1320-7 (1995).
51. T. SAKURAI. N. OHNO and T. YADOMAE, J Leukoc Biol 60 118-24 (1996).