| Title | ANTIOXIDANT PROTECTION OF BIO-NORMALIZER IN CEREBRAL ISCHAEMIA-REPERFUSION INJURY IN THE GERBIL |
|---|---|
| Year | 1993 |
| Author | Librado A. Santiago, James Akira Osato, Norio Ogawa and Akitane Mori |
| Publisher | Neuro Reportv |
Antioxidant Protection of Bio-normalizer in Cerebral Ischaemia-Reperfusion Injury in the Gerbil
Librado A. Santiago, James Akira Osato1, Norio Ogawa and Akitane MoriCA
Department of Neuroscience, Institute of Molecular and Cellular Medicine, Okayama University Medical School, 2-5-1 Shikata-cho, Okayama 700, Japan; 1Sun-O International Inc., Gifu 500, Japan
CACorresponding Author
In this study we used cerebral ischaemia-reperfusion injury (IRI) as an experimental model to analyze the effects of Bio-normalizer (BN, a naturally fermented health food product) on reactive oxygen species related changes in different brain regions of gerbils. Pre-administration of BN solutions (0.1% and 1%) for 45 days produced a significant reduction in IRI-mediated increase in membrane lipid peroxidation, as shown by the decreased carbon-centered radicals and thiobarbituric acid-reactive substances in several brain regions including the cerebral cortex, the hippocampus and the striatum. BN however has no effect on the mitochondrial superoxide dismutase activity.
Keywords: Cerebral ischaemia-reperfusion injury; Hydroxyl radical scavenger; Carbon-centered radicals; Thiobarbituric acid-reactive substances; Superoxide dismutase; Antioxidant; Bio-normalizer
Introduction
Cerebral ischaemia-reperfusion injury (IRI) in the gerbil generates reactive oxygen species (ROS) which cause membrane lipid peroxidation and enzyme inactivation (e.g.loss of glutamine synthetase activity).1-4 Oxidative stress, a major contributory factor in the aetiology of certain cerebrovascular diseases such as cerebral ischaemia is in part attributed to superoxide radical, hydroxyl radical, oxygen- and carbon-centered radicals, and hydrogen peroxide. 5,6 Antoxidants such as vitamin E, glutathione, ascorbate, superoxide dismutase (SOD). catalase, bifemalane hydrochloride, idebenone and indeloxazine hydrochloride, among others, as well as natural herbal medicines with antioxidant properties like alkaloids, lignans and TJ-960 have been analyzed as protective agents in cerebral IRI.6-10
Bio-catalyser a.r No 11 (Bio-normalizer), a commercially available natural health product from a fermented mixture of Carica papaya Linn. (a Philippine medicinal plant), other tropical herbal plants, and cereals from Japanese tropical and traditional foods, exhibits diverse and wide-ranging biological, physiological and therapeutic properties. We have previously reported that Bio-normalizer (BN) potently scavenged hydroxyl radicals, quenched superoxide and 1,1-diphenyl-2-picrylhydrazyl (DPPH) radicals and inhibited the thiobarbituric acid-reactive substances (TBARS) in FeCl3-induced epileptic focus in rats.11 Pre-treating the same experimental rat model of epilepsy with BN decreased the release of serotonergic and dopaminergic neurones in the striatum.12 The mitochondrial and cytosolic SOD activities increased and the TBARS decreased in several brain regions of 116-week old aged rats pre-administered with BN for 27 weeks, which corresponded to the observed increased life span and improved physical conditions of the rats.13
To probe deeper into the ROS scavenging action and antioxidant role of BN in cerebrovascular diseases, we investigated its inhibiting effect on membrane lipid peroxidation presumed to be initiated by hydroxyl radicals as a result of IRI in gerbil brain. Using electron spin resonance (ESR) spin trapping technique and flourometry, we measured the formed lipid carbon-centered (CCR) and TBARS, respectively in selected brain regions to determine the IRI-mediated changes in lipid peroxidation. The changes in SOD activity as shown by its ability to dismutase the generated superoxide radical in vitro was also analyzed through an ESR spectrometer.
Materials and Methods
Test material: BN is a white, sweet, granular, naturally fermented health food product marketed in Japan and in the Philippines by Sun- o International Inc., Gifu, Japan.
Animals and treatment: Experimental animals used were 40 male Mongolian gerbils (70-90 g) categorized into two groups: sham-operated and ischaemic. The sham-operated group was further classified according to diet—one had ad libitum access to standard diet and water (A) and the other, the diet and BN in water (0.1%) in place of pure water (B). The ischaemic group was subdivided into 3 diet groups — the first was given the diet and water (C), the second, the diet and 0.1% BN solution (D), and the third the diet and 1.0% BN solution (E). These dosages approximate the recommended dose for humans (i.e.3-9 g daily). The five subgroups of eight animals per cage were housed at 25ºC and humidity of 50 ± 5% for 45 days. For surgery, the animals were anaesthesized with ketamine hydrochloride (100 mg kg-1 i.p.). Silk threads were placed around both common carotid arteries without interrupting carotid blood flow. On the following day, both arteries were exposed and occluded with surgical clips under light anaesthesia. The clips were released after 5 min to restore the carotid artery blood flow. The sham-operated controls were treated in the same manner without performing the occlusion procedure. The gerbils were sacrificed by decapitation after 30 min of reperfusion (following previous data that a tremendous increase in TBARS occur at this time)14 and the brain was immediately excised. The cortex, cerebellum, pons-medulla oblongata, midbrain, striatum and hippocampus were dissected and homogenized separately in 10 volumes of ice-cold 0.1 M phosphate buffer, pH 7.4.
Measurement of CCR: 0.5 ml of brain homogenate was incubated in 0.05 ml of 2 mM FeCl2-ascorbic acid mixture and 0.45 ml of 0.1.M phosphate buffer at 37ºC for 15 min. To 200 ml of the incubated homogenate was added 2 ml of 5,5’-dimethyl-1-pyrroline-N-oxide. The mixture was vortexed and placed in an ESR spectrometry flat cell (169 ml volume capacity). The spin adducts of DMPO-CCR were recorded 50 s after the addition of DMPO. The number of spin adducts of DMPO-CCR was calculated from the ratio of the signal height intensity of the sample and the standard 2,2,6,6-tetramethyl-4-hydroxylpiperidine-1-oxyl (TEMPOL) having a known spin number. Details are given in a previous report.15
Measurement of TBARS: The method of Ohkawa et al was used.16 Briefly, to 0.1 ml of the homogenized brain tissue was added (a) 0.2 ml of 8.1% sodium dodecyl sulfate, (b) 1.5 ml of 20% sodium acetate solution, pH 3.5, (c) 1.5 ml of 0.8 % sodium thiobarbituric acid (TBA), and (d) 0.7 ml of distilled water; then mixed and incubated at 100ºC in a water bath for 1 h. After cooling, 1 ml of distilled water and 5 ml of n-butanol and pyridine mixture (5:1) were added and shaken vigorously for 1 min using an automatic shaker. After centrifugation at 3,000 rpm for 10 min, the absorbance of the supernatant containing the TBARS was read at Ex 515 nm and Em 532 nm using a flourometer. The values were determined from a standard curve of 1,1,3,3-tetramethoxypropane. The formed lipid peroxide was expressed as nmol TBARS/g tissue wet weight.
Measurement of SOD activity: The procedure of Hiramatsu et al was used. 17 The cerebral cortex homogenate was centrifuged at 1,650 g for 15 min. The supernatant was re-centrifuged at 11,000 g for 30 min and the crude mitochondrial pellet obtained was suspended in phosphate buffer, pH 7.8. This suspension was used as the mitochondrial Mn-SOD. The amount of the resulting supernatant after ultracentrifugation at 105,000 g containing the cytosolic Cu/ZnSOD was not enough for ESR analysis. 50ml of 2mM hypoxanthine, 35 ml of 11 mM diethylenetriaminepentaacetic acid, 50 ml of the mitochondrial pellet suspension, 15 ml of DMPO, and 50 ml of xanthine oxidase [81.6 ml in 5 ml phosphate buffer were vortexed and placed in an ESR spectrometry flat cell. The spin adducts of superoxide radicals (DMPO-OOH) were recorded exactly 60 s after the addition of DMPO. A standard SOD curve was prepared from different dilutions of SOD. Manganese oxide was used as the internal standard. The spin number of the DMPO-OOH was calculated based from the known spin number of the standard TEMPOL. The ESR parameters were the same as in the previous studies. 13,17
Statistical analysis: Data difference between groups was tested for significance using Student’s t-test.
Results
Sham-operated versus ischaemic gerbils: The reaction of Fe2+ and ascorbic aid in the presence of DMPO produced prominent ESR signals which indicated the trapped CCR (data not shown) in the brain tissue homogenate of sham-operated gerbils (control group). The ESR spectrum consists of six-line signals of the spin adducts of DMPO-CCR which is characterized by hyperfine coupling constants from the nitroxide nitrogen (AN) and the b-hydrogen (A11b) nuclei of 15.52 G (where 1 Gauss = 10-4 Tesla) and 23.03 G, respectively. 5 min bilateral carotid occlusion in gerbil produced forebrain ischaemia resulting in significant increases of CCR in the cortex (p<0.05), hippocampus (p<0.025), midbrain (p<0.05) and striatum (p<0.001). On the other hand, TBARS levels were significantly elevated in the cortex (p<0.01) hippocampus (p<0.01) and midbrain (p<0.005) and apparently increased in the striatum although not significantly. No difference was observed between BN-treated sham-operated gerbils and untreated sham-operated with regard to CCR and TBARS in most brain parts with the exception of a marked reduction in TBARS in the midbrain of BN-treated gerbils (Figs 1 and 2). On the other hand the TBARS in the cerebellum of ischaemic gerbils pre-administered with 0.1% and 1% BN significantly decreased compared with untreated sham-operated gerbils (p<0.01) and BN-treated sham-operated gerbils (p<0.01 and p<0.025) (Fig. 2).
Ischaemic (control) versus BN-treated ischaemic gerbils: Following 5 min hypoxia and 30 min reoxygenation, the CCR were significantly inhibited in the midbrain (p<0.05), hippocampus (p<0.001 or p<0.01), and striatum (p<0.005 or p<0.001) in reperfused gerbils pre-orally administered with 0.1% and 1.0% BN solutions for 45 days. The TBARS were also significantly inhibited in the cortex (p<0.05 or p<0.025), midbrain (p<0.05), hippocampus (p<0.05) and striatum (p<0.05) (Figs. 1 and 2).
Endogenous SOD in gerbils: ESR spin trapping of superoxide radical with DMPO was conducted using the hypoxanthine-xanthine oxidase system. The degredation rate of the spin adducts of superoxide radical (DMPO-OOH) by SOD standard was used in the calculation of SOD activity in the different brain regions. ESR hyperfine splitting constants for DMPO-OOH spin adducts are AN = 13.7 G, AHb = 11.8 G and AHg = 2.1 G. The mitochondrial SOD activity showed a slight decline during IRI in gerbil cortex, although insignificant (Fig. 3). Pre-administration of BN solution (0.1% and 1.0%) for 45 days brought about no significant change on the mitochondrial SOD in the cerebral cortex of sham-operated and ischaemic gerbils (Fig. 3).
Discussion
When ischaemic tissues are reperfused the resulting superoxide radical (from the reaction between xanthine oxidase and xanthine or hypoxanthine) could be a key intermediate in IRI damage; and in the presence of hydrogen peroxide and catalytic metal ions such as iron and copper through Fenton chemistry may generate hydroxyl radical in the brain.1-6 The hydroxyl radical initiates the process of lipid peroxidation by abstracting a hydrogen atom from polyunsaturated fatty acid side chain in membrane phospholipids. Abstraction of a hydrogen atom leaves behind CCR in the membrane. The oxidative process proceeds to form a peroxyl radical and finally gets converted into lipid hydroperoxides.5,6 The extent of membrane lipid peroxidation could be measured in terms of CCR and TBARS.
Our results demonstrate that membrane lipid peroxidation indeed occurred during IRI in gerbil brain causing the increased CCR and TBARS levels in the forebrain regions. The most affected regions in the gerbil are the cerebral cortex, hippocampus, striatum and midbrain. Perhaps due to regional differences, metal-ion release and variant antioxidant and antioxidant enzyme contents, the reperfused brain tissues tended to show different sensitivities to ROS-induced damage during IRI. Pre-administration of BN in gerbils produced a significant reduction in IRI-mediated increase in lipid peroxidation as shown by the decreased CCR and TBARS in several brain regions including the cerebral cortex, the hippocampus, the striatum and the midbrain. These data are in congruence with previous findings on BN’s ability to inhibit TBARS iniron-induced epileptic rats11 and in 116-week old aged rats.13
Another possible contributing factor in IRI damage is the inactivation of glutamine synthetase, a major enzyme for glutamate removal that allows glutamate accumulation in the brain.4 Glutamate and aspartate are excitatory neurotransmitters implicated in epileptic seizures that cause neurones to fire off continuously until they are damaged, impairing neurotrasmission.18,19 Using, an iron-injected model of epileptic rats we have demonstrated that pre-treatment with BN brought about a significant reduction in the intrastriatal release of monoamines and their metabolites.12 It was speculated that the increase in biogenic amines in iron-induced epileptic rats may be due to the excitatory release of amino acids such as aspartate. Recent data indicate that BN has some inhibiting ability on potassium-stimulated release of [3H] aspartate in the hippocampal slices of ddY mice (unpublished) suggesting neuroprotective effects against epileptic seizures and IRI. Exogenously supplied SOD but not endogenous SOD has some minimizing effects on brain injury after ischaemia-reperfusion in animals.4 Corollarily, the administration of recombinant human SOD can reduce the delayed vasogenic oedema developing at 3 h of reperfusion.20 We found that neither the endogenous mitochondrial SOD in the cerebral cortex of gerbils nor the exogenously pre-administered BN afforded protection during IRI. This may be due to the fact that BN has weak scavenging action on superoxide radicals11 and is a crude naturally fermented health food product. The 45 day administration of BN seemed short to counteact the oxidative damage during IRI in gerbils. The six-month pre-administration of BN in 116-week old rats however, produced a remarkable increase in the mitochondrial and cytosolic SOD activities in the cerebral cortex, pons-medulla oblongata and hippocampus.13
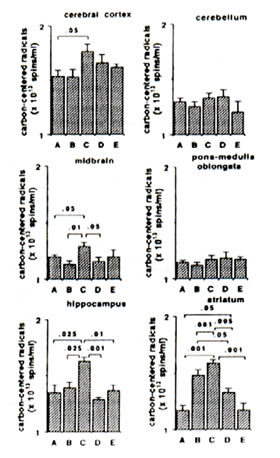
FIG. 1. Effect of Bio-normalizer on carbon-centered radicals during ischaemia-reperfusion injury in gerbil brain. A: sham-operated controls given food and water, B: sham-operated controls given food and 0.1% Bio-normalizer, C: cerebral ischaemic gerbils given food and water, D: as in C but given food and 0.1% Bio-normalizer, E: as in C but given food and 1.0% Bio-normalizer for 45 days. Data are expressed as the mean ± s.e.m. of 5-7 gerbils and extracted from the ESR spectrum of spin adducts of carbon-centered radicals having hyperfine splitting constants AN =15.52 G and AHb =23.03 G.
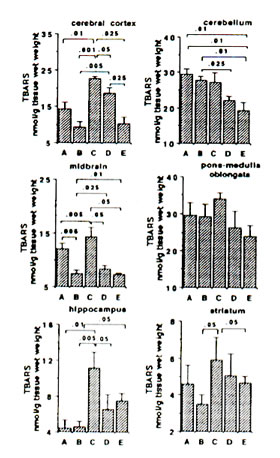
FIG. 2. Effect of Bio-normalizer on thiobarbituric acid-reactive substances (TBARS) during ischaemia-reperfusion injury in gerbil brain. A: sham-operated controls given food and water, B: sham-operated controls given food and 0.1% Bio-normalizer, C: cerebral ischaemic gerbils administered with food and water, D: as in C but given food and 0.1% Bio-normalizer, E: as in C but given food and 1.0% Bio-normalizer for 45 days. Data are expressed as the mean ± s.e.m. of 5-7 gerbils.
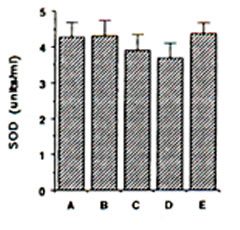
FIG. 3. Effect of Bio-normalizer on mitichondrial superoxide dismutase during ischaemia-reperfusion injury in gerbil brain. A: sham-operated controls given food and water, B: sham-operated controls given food and 0.1% Bio-normalizer, C: cerebral ischaemic gerbils given food and water, D: as in C but given food and 0.1% Bio-normalizer, E: as in C but given food and 1.0% Bio-normalizer for 45 days. Data are expressed as the mean ± s.e.m. of 5-7 gerbils and extracted from the ESR spectrum of spin adducts of superoxide radicals (DMPO-OOH) having hyperfine splitting constants of AN = 13.7 G, AHb = 11.8 G, AHg and = 2.1G.
Several reports show that brief (5 min) bilateral carotid occlusion in the gerbil produces forebrain ischaemia resulting in almost complete neuronal loss21,22 and delayed neuronal cell death in the CA1 region of hippocampus.14,23,24 Usually, these transient ischaemic attacks serve as ominous warning signs of impending stroke and cerebral infarction.25 By reducing significantly IRI-induced damage on membrane lipid peroxidation, in particular in the hippocampus, it is likely that BN could act as a neuroprotective agent against delayed CA1 hippocampal neuronal death
Conclusion
This study shows ESR evidence for the role of ROS in cerebrovascular diseases such as cerebral ischaemia in the gerbil. Pre-administration of BN in the animals produced a significant reduction in membrane lipid peroxidation as shown by the decreased CCR and TBARS levels in the cerebral cortex, midbrain, hippocampus and striatum. The results suggest the BN (through its antioxidant and free radical scavenging activities) affords significant cerebroprotection in the gerbil model of transient forebrain ischaemia.
References
- Cao W., Carney JM, Duchon A et al. Neurosci Lett 88,233-238(1988).
- Watson BD and Ginsberg MD. Annals NY Acad Sci 559,269-281(1989).
- Floyd RA. FASEB J 4, 2587-2597 (1990).
- Oliver CN, Stark-Reed PE, Stadtman ER et al. Proc Natl Acad Sci USA 87,5144 –5147 (1990).
- Halliwell B. J Neurochem 59,1609-1623 (1992).
- Packer L. Free radical scavengers and antioxidants in prophylaxis and treatment of brain diseases. In: Packer L. Prilipko L, Christen Y, eds. Free Radicals in the Brain. Springer-Verlag/Berlin, 1992:1-20.
- Ogawa N, Haba K, Yoshikawa H et al. Res Comms Chem Pathol Pharmacol 61,285-288(1988).
- Izumiyama K and Kogure K. J Cerebr Blood Flow Metab 1, Suppl. 9.S 187 (1989).
- Xue JY, Liu GT, Wei HL et al. Free Radical Biol Med 12, 127-135 (1992).
- Haba K, Ogawa N and Mori A. Neurochem Res 15, 487-493 (1990).
- Santiago LA, Osato JA, Hiramatsu M et al. Free Radical Biol Med 11,379-383 (1991).
- Santiago LA, Osato JA, Kabuto H et al. Med Sci Res 21, 139-141 (1993).
- Santiago LA, Osato JA, Liu J et al. Neurochem Res 18, 711-717 (1993).
- Haba K, Ogawa N, Mizukawa K et al. Brain Res 540, 116-122 (1991).
- Santiago LA, Hiramatsu M And Mori A. J Nutr Sci Vitam 38, 297-304 (1992).
- Ohkawa H, Ohishi S and Yagi K. Anal Biochem 95, 351-358 (1979).
- Hiramatsu M, Kohno M, Edamatsu R et al. J Neurochem 58, 1160-1164 (1992).
- Pelligrini-Giampietro DE, Cherici G, Alesiani M et al. J Neurosci 10, 1035-1041 (1988).
- Benveniste H,Drejer J, Schousboe A et al. J Neurochem 43, 1369-1374 (1984).
- Uyama O, Shiratsuki N, Matsuyama T et al. Free radical Biol Med 8, 265-268 (1990).
- Nakano S, Kogure K and Fujikura H. Neuroscience 38,115-124 (1990)
- Pulsinelli WA, Brierley JB and Plum F. Ann Neurol 11, 491-498 (1982).
- Kirino T. Brain Res 239. 57-69 (1982).
- Kitagawa K, Matsumoto M, Oda T et al. Neuroscience 35,551-558 (1990).
- Evans GW, Howard G, Murros KE et al. Stroke 22, 431-436 (1991).
Received 22 March 1993;
Accepted 7 June 1993